Insulinoma and pheochromocytoma are rare neuroendocrine tumors that significantly impact hormone regulation, causing symptoms such as hypoglycemia and hypertension, respectively. Accurate diagnosis and effective treatment are critical for managing these conditions and preventing serious complications. Explore the full article to understand their differences, symptom management, and advanced treatment options tailored for your health.
Table of Comparison
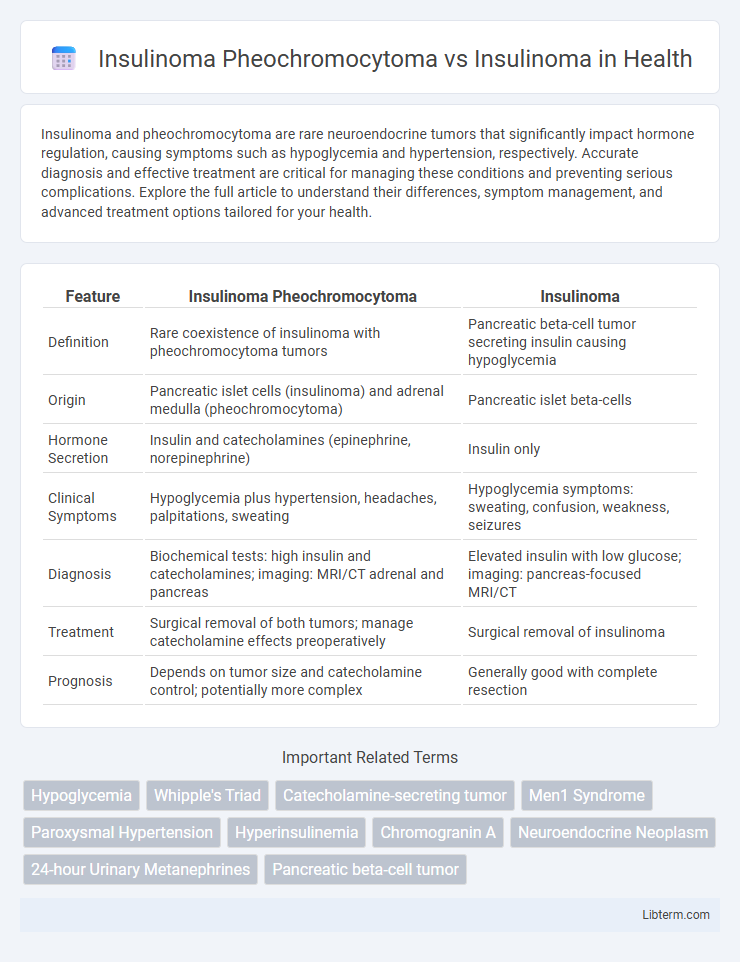
| Feature | Insulinoma Pheochromocytoma | Insulinoma |
|---|---|---|
| Definition | Rare coexistence of insulinoma with pheochromocytoma tumors | Pancreatic beta-cell tumor secreting insulin causing hypoglycemia |
| Origin | Pancreatic islet cells (insulinoma) and adrenal medulla (pheochromocytoma) | Pancreatic islet beta-cells |
| Hormone Secretion | Insulin and catecholamines (epinephrine, norepinephrine) | Insulin only |
| Clinical Symptoms | Hypoglycemia plus hypertension, headaches, palpitations, sweating | Hypoglycemia symptoms: sweating, confusion, weakness, seizures |
| Diagnosis | Biochemical tests: high insulin and catecholamines; imaging: MRI/CT adrenal and pancreas | Elevated insulin with low glucose; imaging: pancreas-focused MRI/CT |
| Treatment | Surgical removal of both tumors; manage catecholamine effects preoperatively | Surgical removal of insulinoma |
| Prognosis | Depends on tumor size and catecholamine control; potentially more complex | Generally good with complete resection |
Introduction to Insulinoma and Pheochromocytoma
Insulinoma is a rare neuroendocrine tumor of the pancreas that excessively secretes insulin, leading to hypoglycemia characterized by symptoms such as confusion, sweating, and palpitations. Pheochromocytoma is a catecholamine-secreting tumor arising from adrenal medulla chromaffin cells, causing episodic hypertension, headaches, and tachycardia through excessive release of adrenaline and noradrenaline. Both tumors are part of the multiple endocrine neoplasia syndromes but differ significantly in origin, hormonal activity, and clinical presentation.
Overview of Insulinoma
Insulinoma is a rare neuroendocrine tumor arising from pancreatic beta cells, characterized by excessive insulin secretion leading to hypoglycemia. Unlike pheochromocytoma, which originates from adrenal medulla chromaffin cells and produces catecholamines, insulinomas primarily affect glucose metabolism and cause symptoms such as confusion, sweating, and weakness due to low blood sugar. Diagnosis of insulinoma relies on supervised fasting tests, blood glucose, insulin, C-peptide levels, and imaging studies like endoscopic ultrasound or MRI to localize the tumor.
Overview of Pheochromocytoma
Pheochromocytoma is a rare neuroendocrine tumor originating from chromaffin cells of the adrenal medulla, characterized by excessive catecholamine secretion leading to episodic hypertension, palpitations, and headaches. Unlike insulinomas, which primarily cause hypoglycemia due to inappropriate insulin secretion, pheochromocytomas induce symptoms related to adrenergic overstimulation. Diagnosis relies on biochemical assays measuring elevated plasma metanephrines and imaging studies such as MRI or CT scans for tumor localization.
Pathophysiological Differences
Insulinoma is a pancreatic neuroendocrine tumor characterized by excessive insulin secretion causing hypoglycemia, primarily due to autonomous beta-cell proliferation. Pheochromocytoma arises from chromaffin cells of the adrenal medulla, secreting catecholamines like norepinephrine and epinephrine, leading to hypertension and metabolic disturbances. The key pathophysiological difference lies in insulinoma's direct impact on glucose homeostasis through insulin overproduction, whereas pheochromocytoma alters metabolic pathways via catecholamine excess without affecting insulin secretion.
Clinical Presentation and Symptoms
Insulinomas and pheochromocytomas differ significantly in clinical presentation; insulinomas primarily cause hypoglycemia characterized by symptoms such as confusion, sweating, weakness, and palpitations resulting from excessive insulin secretion. Pheochromocytomas present with episodic hypertension, headaches, sweating, and palpitations due to catecholamine hypersecretion. While insulinomas manifest with neuroglycopenic and adrenergic symptoms related to low blood glucose levels, pheochromocytomas mainly produce cardiovascular symptoms related to catecholamine excess.
Diagnostic Approaches: Insulinoma vs Pheochromocytoma
Insulinoma diagnosis primarily involves measuring fasting insulin and C-peptide levels alongside a supervised 72-hour fast to confirm endogenous hyperinsulinism. Pheochromocytoma diagnosis relies on biochemical testing such as plasma-free metanephrines or 24-hour urinary fractionated metanephrines to detect catecholamine excess. Imaging modalities like CT, MRI, and functional PET scans further differentiate tumor localization and are critical for both insulinoma and pheochromocytoma identification.
Laboratory Findings and Biomarkers
Insulinoma pheochromocytoma presents with elevated plasma metanephrines and normetanephrines, highlighting catecholamine excess, whereas insulinoma shows markedly increased insulin, C-peptide, and proinsulin levels during hypoglycemia. Laboratory findings in insulinoma pheochromocytoma often reveal concurrent hypoglycemia and hypertension due to secreted catecholamines, contrasting with isolated hypoglycemia in insulinoma. Biomarkers such as chromogranin A may be elevated in both, but the distinct catecholamine metabolites differentiate insulinoma pheochromocytoma from pure insulinoma.
Imaging Modalities for Tumor Localization
Imaging modalities for tumor localization in insulinoma and pheochromocytoma differ significantly due to their distinct tissue origins and metabolic characteristics. Insulinomas are primarily localized using high-resolution multiphase contrast-enhanced CT, MRI with T1 and T2 sequences, and functional imaging like 68Ga-DOTATATE PET/CT, which targets somatostatin receptors expressed by neuroendocrine tumors. Pheochromocytomas are best identified with 123I-MIBG scintigraphy and 18F-FDOPA PET/CT, leveraging their high catecholamine synthesis, alongside anatomical imaging from MRI and CT to assess tumor size and adrenal involvement.
Treatment Strategies and Prognosis
Treatment strategies for insulinoma involve surgical resection as the primary approach, often leading to excellent prognosis with high cure rates. Pheochromocytoma associated with insulinoma, especially in multiple endocrine neoplasia type 1 (MEN1) syndrome, requires preoperative alpha-adrenergic blockade followed by adrenalectomy to control catecholamine excess and prevent intraoperative complications. Prognosis differs significantly; isolated insulinomas generally have favorable outcomes post-surgery, whereas insulinoma-pheochromocytoma cases require multidisciplinary management due to potential malignancy and recurrence risks.
Key Differences and Clinical Implications
Insulinoma pheochromocytoma syndrome involves concurrent tumors of the pancreas and adrenal medulla, characterized by insulin-secreting pancreatic neuroendocrine tumors and catecholamine-producing adrenal tumors, whereas isolated insulinoma refers solely to pancreatic beta-cell tumors causing hypoglycemia. The dual pathology of insulinoma pheochromocytoma complicates clinical management due to fluctuating glucose and catecholamine levels, increasing risks of hypertensive crises alongside hypoglycemic episodes. Accurate differentiation through biochemical assays and imaging is crucial for tailored surgical intervention and perioperative care to mitigate life-threatening metabolic and cardiovascular complications.
Insulinoma Pheochromocytoma Infographic