Sarcoidosis is an inflammatory disease characterized by the formation of tiny clumps of inflammatory cells called granulomas in various organs, most commonly the lungs and lymph nodes. Symptoms can vary widely, including fatigue, shortness of breath, and persistent cough, making early diagnosis challenging but crucial for effective management. Explore the rest of the article to understand sarcoidosis symptoms, treatments, and how you can manage this complex condition.
Table of Comparison
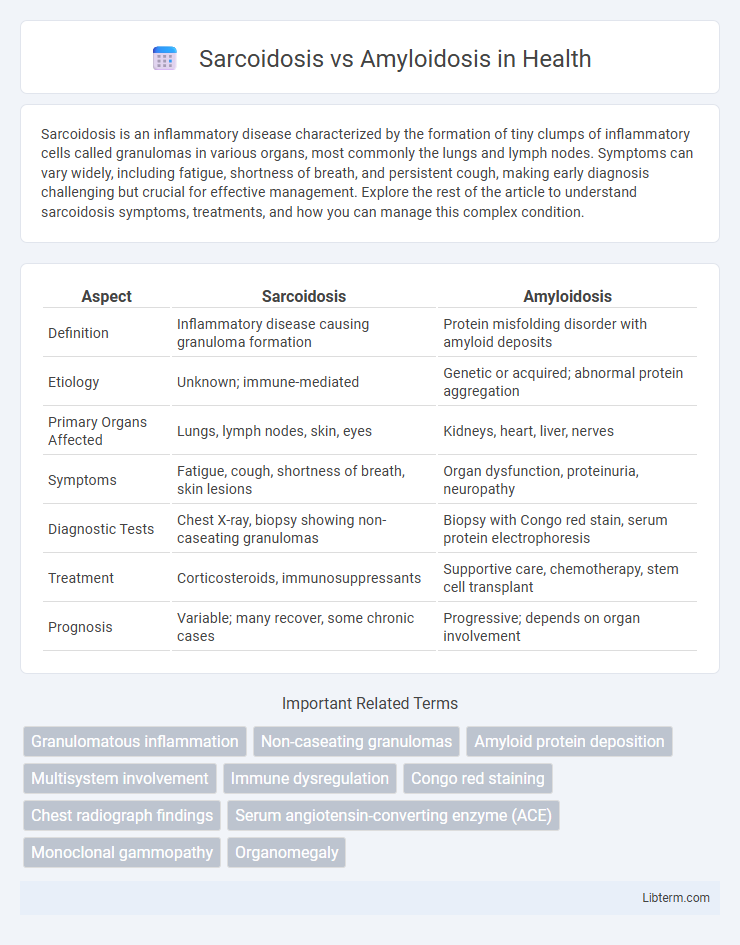
| Aspect | Sarcoidosis | Amyloidosis |
|---|---|---|
| Definition | Inflammatory disease causing granuloma formation | Protein misfolding disorder with amyloid deposits |
| Etiology | Unknown; immune-mediated | Genetic or acquired; abnormal protein aggregation |
| Primary Organs Affected | Lungs, lymph nodes, skin, eyes | Kidneys, heart, liver, nerves |
| Symptoms | Fatigue, cough, shortness of breath, skin lesions | Organ dysfunction, proteinuria, neuropathy |
| Diagnostic Tests | Chest X-ray, biopsy showing non-caseating granulomas | Biopsy with Congo red stain, serum protein electrophoresis |
| Treatment | Corticosteroids, immunosuppressants | Supportive care, chemotherapy, stem cell transplant |
| Prognosis | Variable; many recover, some chronic cases | Progressive; depends on organ involvement |
Introduction to Sarcoidosis and Amyloidosis
Sarcoidosis is an inflammatory disease characterized by the formation of non-caseating granulomas in multiple organs, most commonly the lungs and lymph nodes, leading to tissue inflammation and impaired organ function. Amyloidosis involves the extracellular deposition of misfolded amyloid proteins in various tissues, causing organ dysfunction due to progressive amyloid fibril accumulation primarily affecting the heart, kidneys, and nervous system. Both diseases present complex multisystem challenges but differ fundamentally in their pathophysiology, with sarcoidosis driven by immune dysregulation and amyloidosis by protein misfolding and aggregation.
Definition and Overview
Sarcoidosis is an inflammatory disease characterized by the formation of non-caseating granulomas primarily in the lungs and lymph nodes. Amyloidosis involves the abnormal deposition of amyloid proteins in tissues and organs, leading to impaired function. Both conditions affect multiple organ systems but differ fundamentally in their pathological mechanisms and clinical presentations.
Causes and Risk Factors
Sarcoidosis is caused by an abnormal immune response leading to granuloma formation, often triggered by genetic predisposition and environmental factors such as exposure to dust, mold, or certain infections. Amyloidosis results from abnormal protein aggregation, often linked to chronic inflammatory diseases, genetic mutations, or conditions like multiple myeloma. Risk factors for sarcoidosis include age 20-40, African American ethnicity, and family history, while amyloidosis risk increases with age, chronic kidney disease, and hereditary factors.
Pathophysiology: How the Diseases Develop
Sarcoidosis develops through an exaggerated immune response leading to the formation of non-caseating granulomas primarily in the lungs and lymph nodes, driven by T-helper 1 (Th1) cells and macrophage activation. Amyloidosis results from the extracellular deposition of misfolded amyloid proteins, either light chains in AL amyloidosis or serum amyloid A in AA amyloidosis, causing organ dysfunction through structural disruption. The pathophysiological distinction lies in sarcoidosis's granulomatous inflammation versus amyloidosis's protein aggregation and tissue infiltration.
Clinical Manifestations and Symptoms
Sarcoidosis commonly presents with bilateral hilar lymphadenopathy, pulmonary infiltrates, and skin lesions such as erythema nodosum, often accompanied by fatigue, cough, and dyspnea. Amyloidosis manifests through organ-specific symptoms depending on amyloid deposition, including nephrotic syndrome, restrictive cardiomyopathy, hepatomegaly, and peripheral neuropathy, with characteristic signs like macroglossia and easy bruising. Both diseases may involve multisystem complications, but sarcoidosis features granulomatous inflammation, whereas amyloidosis is marked by extracellular amyloid protein deposits disrupting tissue function.
Diagnostic Criteria and Testing
Sarcoidosis diagnosis relies on clinical presentation, chest X-rays revealing bilateral hilar lymphadenopathy, and tissue biopsy showing non-caseating granulomas, supported by elevated serum angiotensin-converting enzyme (ACE) levels. Amyloidosis diagnosis involves Congo red staining of tissue biopsies demonstrating amyloid fibril deposits with apple-green birefringence under polarized light, alongside serum and urine protein electrophoresis to detect abnormal light chains, especially in AL amyloidosis. Advanced imaging such as cardiac MRI aids in identifying organ involvement for both conditions.
Organ Systems Commonly Affected
Sarcoidosis primarily affects the lungs, lymph nodes, skin, and eyes, with granulomas forming in these tissues, leading to respiratory symptoms, skin lesions, and vision problems. Amyloidosis involves abnormal protein deposits in multiple organs, commonly targeting the heart, kidneys, liver, and nervous system, resulting in organ dysfunction and heart failure, nephrotic syndrome, or neuropathy. Both diseases can affect multiple organ systems but differ in pathological mechanisms and predominant sites of involvement.
Treatment Options and Management
Sarcoidosis treatment primarily involves corticosteroids such as prednisone to reduce inflammation and manage symptoms, with immunosuppressive agents like methotrexate used in refractory cases. Amyloidosis management depends on the type; AL amyloidosis often requires chemotherapy, including proteasome inhibitors like bortezomib, while ATTR amyloidosis may be treated with tafamidis or liver transplantation to stabilize or reduce amyloid deposits. Both conditions necessitate regular monitoring of organ function, and supportive care including symptom relief and management of organ-specific complications is crucial.
Prognosis and Long-Term Outcomes
Sarcoidosis typically has a variable prognosis, with many patients experiencing spontaneous remission or manageable chronic disease, while severe cases can lead to organ damage, especially affecting the lungs and heart. Amyloidosis generally carries a poorer long-term outlook due to progressive organ dysfunction caused by amyloid protein deposits, often resulting in heart failure or kidney failure if untreated. Early diagnosis and targeted therapies significantly influence survival rates and quality of life in both conditions.
Key Differences and Comparative Summary
Sarcoidosis is a granulomatous disease characterized by non-caseating granulomas primarily affecting the lungs and lymph nodes, while amyloidosis involves abnormal extracellular deposition of amyloid proteins in various organs, leading to tissue dysfunction. Sarcoidosis often presents with bilateral hilar lymphadenopathy and elevated serum angiotensin-converting enzyme (ACE) levels, whereas amyloidosis diagnosis relies on tissue biopsy with Congo red staining showing apple-green birefringence under polarized light. Treatment for sarcoidosis typically includes corticosteroids to reduce inflammation, while amyloidosis management depends on the amyloid type, often requiring chemotherapy or organ-specific interventions.
Sarcoidosis Infographic