Cystic fibrosis is a genetic disorder that primarily affects the lungs and digestive system, causing thick, sticky mucus buildup that leads to chronic infections and difficulty breathing. Advances in treatment options have significantly improved life expectancy and quality of life for those living with this condition. Discover how you can manage symptoms and support your health by reading the rest of this article.
Table of Comparison
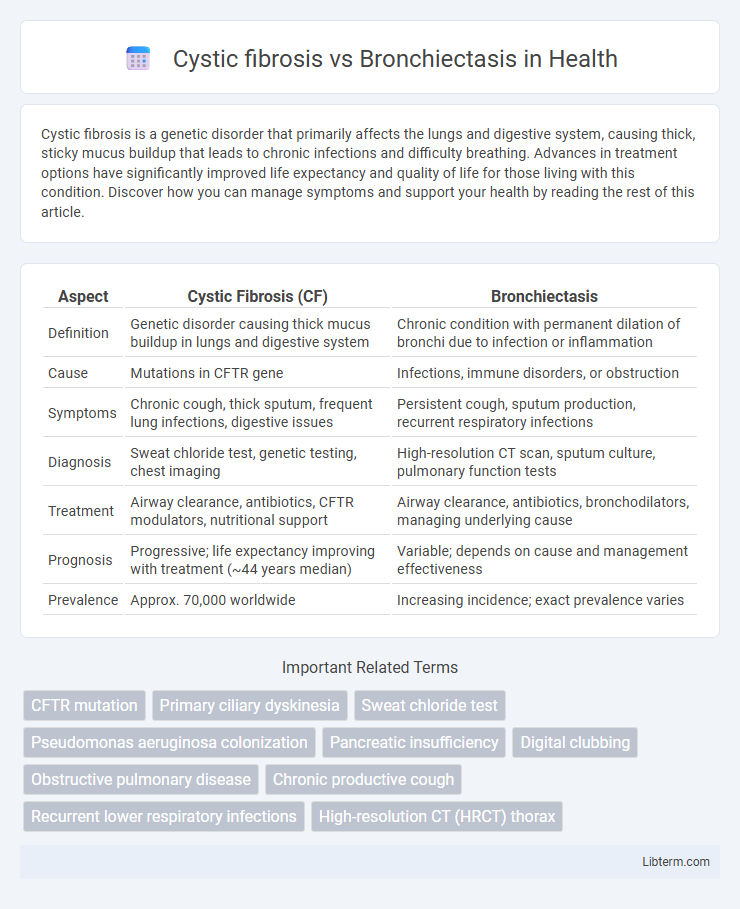
| Aspect | Cystic Fibrosis (CF) | Bronchiectasis |
|---|---|---|
| Definition | Genetic disorder causing thick mucus buildup in lungs and digestive system | Chronic condition with permanent dilation of bronchi due to infection or inflammation |
| Cause | Mutations in CFTR gene | Infections, immune disorders, or obstruction |
| Symptoms | Chronic cough, thick sputum, frequent lung infections, digestive issues | Persistent cough, sputum production, recurrent respiratory infections |
| Diagnosis | Sweat chloride test, genetic testing, chest imaging | High-resolution CT scan, sputum culture, pulmonary function tests |
| Treatment | Airway clearance, antibiotics, CFTR modulators, nutritional support | Airway clearance, antibiotics, bronchodilators, managing underlying cause |
| Prognosis | Progressive; life expectancy improving with treatment (~44 years median) | Variable; depends on cause and management effectiveness |
| Prevalence | Approx. 70,000 worldwide | Increasing incidence; exact prevalence varies |
Overview of Cystic Fibrosis
Cystic fibrosis is a genetic disorder caused by mutations in the CFTR gene, leading to thick, sticky mucus accumulation primarily in the lungs and digestive system. This condition results in chronic respiratory infections, inflammation, and progressive lung damage, often resembling bronchiectasis, which is characterized by permanent airway dilation due to repeated infections or inflammation. Unlike bronchiectasis, which can have multiple causes, cystic fibrosis is specifically linked to inherited CFTR dysfunction affecting ion transport across epithelial cells.
Overview of Bronchiectasis
Bronchiectasis is a chronic lung condition characterized by permanent dilation and damage of the bronchi, leading to persistent cough, sputum production, and recurrent respiratory infections. Unlike cystic fibrosis, which is a genetic disorder causing thick mucus and multisystem involvement, bronchiectasis may arise from various causes such as infections, immune deficiencies, or airway obstructions. Management of bronchiectasis centers on airway clearance, infection control, and preventing disease progression to improve lung function and quality of life.
Key Differences in Etiology
Cystic fibrosis is a genetic disorder caused by mutations in the CFTR gene, leading to thick mucus production and impaired chloride ion transport. Bronchiectasis results from chronic infections or inflammatory conditions that cause permanent dilation of the bronchi, often secondary to factors like recurrent pneumonia or autoimmune diseases. Unlike cystic fibrosis's inherited origin, bronchiectasis primarily arises from acquired damage to the airway structure.
Genetic vs. Acquired Factors
Cystic fibrosis is a genetic disorder caused by mutations in the CFTR gene, leading to defective chloride ion transport and thick mucus production primarily affecting the lungs and digestive system. Bronchiectasis, in contrast, is often an acquired condition resulting from repeated lung infections, immune system disorders, or environmental exposures that cause permanent dilation and damage to the bronchial walls. While cystic fibrosis manifests from inherited genetic mutations, bronchiectasis arises due to external factors and chronic airway inflammation.
Symptoms Comparison
Cystic fibrosis symptoms primarily include persistent cough with thick mucus, frequent lung infections, and difficulty breathing due to airway obstruction. Bronchiectasis symptoms overlap with chronic cough and sputum production but are often distinguished by recurrent chest infections and a history of underlying conditions causing airway damage. Both conditions exhibit respiratory distress and airflow limitation, though cystic fibrosis typically involves multisystem effects beyond the lungs.
Diagnostic Techniques
Diagnostic techniques for cystic fibrosis primarily include sweat chloride tests, genetic testing for CFTR mutations, and chest imaging such as high-resolution CT scans to evaluate lung damage. Bronchiectasis diagnosis relies heavily on high-resolution CT scans to detect bronchial wall thickening and dilatation, alongside sputum cultures to identify infections and pulmonary function tests to assess airflow obstruction. Both conditions require detailed pulmonary evaluation, but cystic fibrosis diagnosis emphasizes genetic and sweat testing, whereas bronchiectasis diagnosis focuses on radiological and microbiological assessments.
Complications and Prognosis
Cystic fibrosis often leads to severe respiratory complications such as chronic lung infections, bronchiectasis, and respiratory failure, with a prognosis that has improved significantly due to advances in gene-targeted therapies, resulting in a median survival exceeding 40 years. Bronchiectasis, frequently caused by recurrent infections or underlying conditions, can result in persistent cough, hemoptysis, and progressive lung function decline, with prognosis varying widely based on etiology and effective management of underlying causes and infections. Both conditions require vigilant monitoring to prevent exacerbations, but cystic fibrosis typically involves a multisystem impact that complicates overall patient outcomes.
Treatment Strategies
Treatment strategies for cystic fibrosis primarily include airway clearance techniques, inhaled antibiotics, and pancreatic enzyme replacement to manage thick mucus and persistent infections. Bronchiectasis treatment focuses on controlling infections with prolonged antibiotic courses, airway clearance, and addressing underlying causes such as immune deficiencies or post-infectious damage. Both conditions benefit from individualized physiotherapy and monitoring to reduce exacerbations and improve lung function.
Quality of Life Considerations
Cystic fibrosis and bronchiectasis both significantly impact respiratory function, leading to chronic cough, sputum production, and recurrent infections, which reduce quality of life through persistent symptoms and frequent hospitalizations. Management strategies, including airway clearance techniques, nutritional support, and tailored pharmacotherapy, aim to improve lung function and minimize exacerbations, directly enhancing patients' physical well-being and daily activities. Psychological support is essential as chronic illness in both conditions often leads to anxiety and depression, further influencing overall quality of life outcomes.
Clinical Management and Long-term Outcomes
Clinical management of cystic fibrosis involves aggressive airway clearance, inhaled antibiotics, and pancreatic enzyme replacement to address multisystem complications, while bronchiectasis treatment focuses on identifying and managing underlying causes, airway clearance, and targeted antibiotic therapy for exacerbations. Long-term outcomes in cystic fibrosis are heavily influenced by early diagnosis through newborn screening, adherence to multidisciplinary care, and advanced therapies like CFTR modulators, resulting in improved survival and quality of life. Bronchiectasis outcomes depend on disease etiology, frequency of exacerbations, and comorbidities, with tailored management improving lung function preservation and reducing hospitalization rates.
Cystic fibrosis Infographic